Product Development
At Tyche MedTech, Inc., we have established (defined, documented and implemented) and maintain a product development process procedure and other related procedures per the FDA-required Design Control in order to ensure that customer requirements are satisfactorily met. The product development process relies on concurrent development activities and entails a fast and accurate product development cycle with five phases which are as follows: (1) Concept Phase, (2) Specification Phase, (3) Development Phase, (4) Pilot-Production Phase, and (5) Commercialization Phase.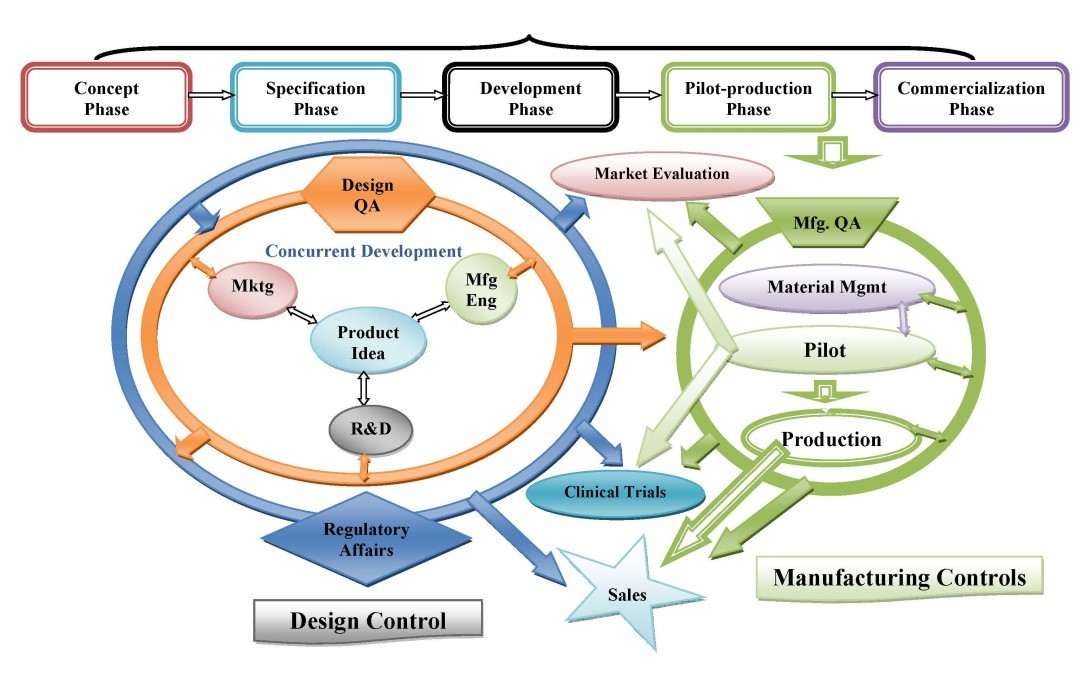
Design Control
At Tyche MedTech, Inc.., we have established (defined, documented and implemented) design control which is an interrelated set of practices and procedures that are incorporated into the design and development process, i.e., a system of checks and balances. Design control makes systematic assessment of the design an integral part of development. As a result, any deficiencies in design input requirements and any discrepancies between the proposed designs and requirements are made evident and corrected earlier in the development process. Design control increases the likelihood that the design transferred to production will translate into a product that is appropriate for its intended use. Design control applies to all changes to the product or manufacturing process design, including those occurring long after a product has been introduced to the market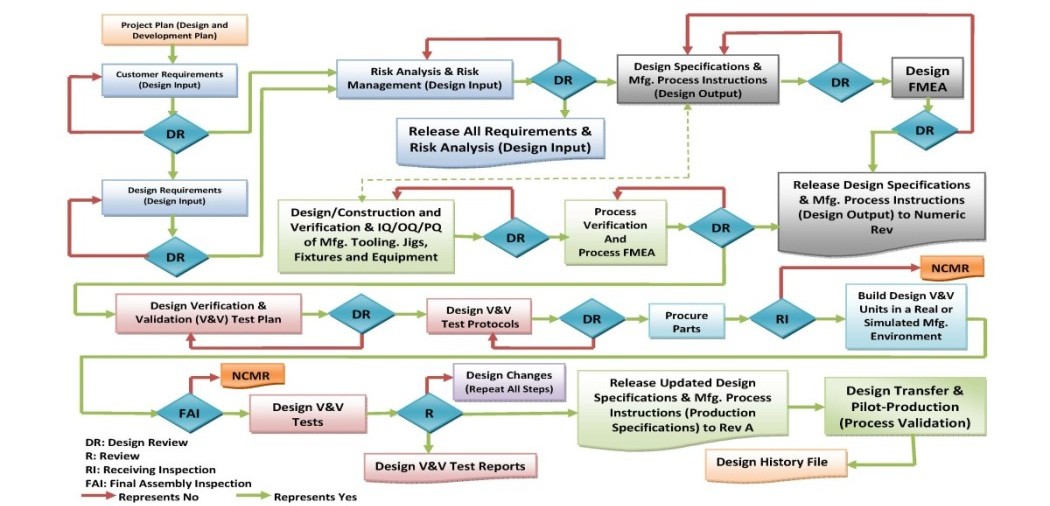
Management Control
At Tyche MedTech, Inc., we have established (defined, documented and implemented) a management control process in order to provide adequate resources for product design, manufacturing, quality assurance, distribution, installation, and servicing (if applicable) activities; assure the quality management system is functioning properly; monitor the quality management system; and make necessary adjustments. A quality management system that has been implemented effectively and is monitored to identify and address problems is more likely to produce products that function as intended. Also our management with executive responsibility ensures that an adequate and effective quality management system has been established.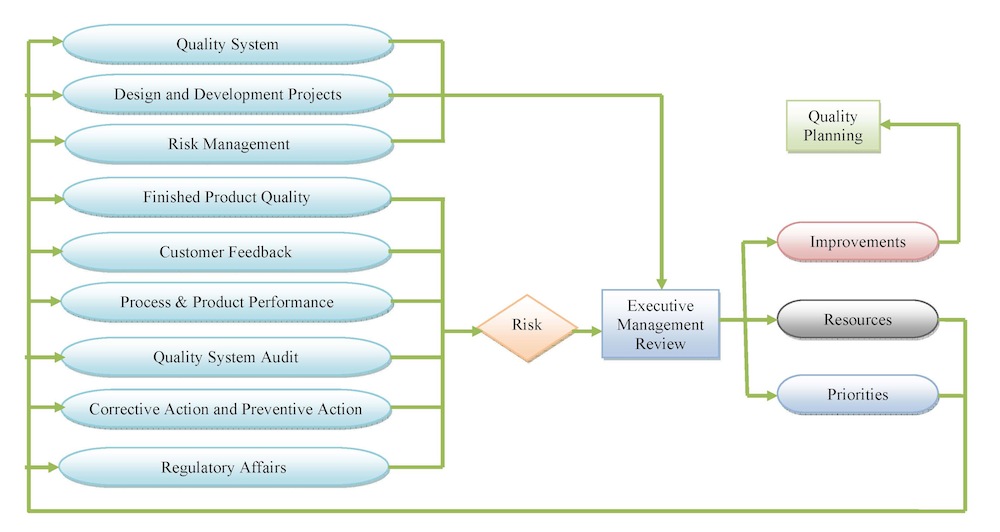
Document Control
At Tyche MedTech, Inc., we have established (defined, documented and implemented) a procedure for a system of document and data control to ensure that medical product documentation is created, approved, distributed, maintained and revised in a controlled manner.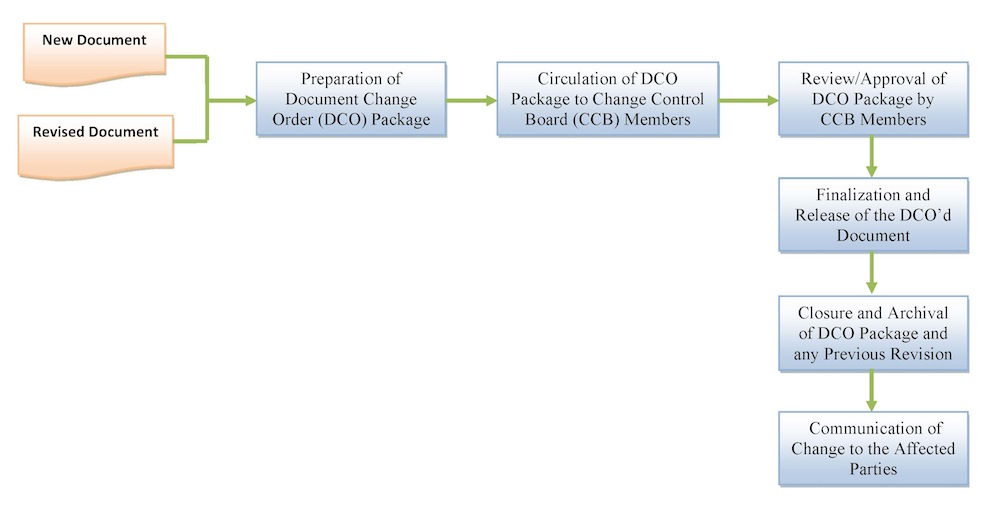
Purchasing Control
At Tyche MedTech, Inc., we have defined, documented and implemented controlled purchasing procedures that will ensure that purchased components, materials, contractors, parts and products conform to requirements and that these requirements are properly understood by suppliers.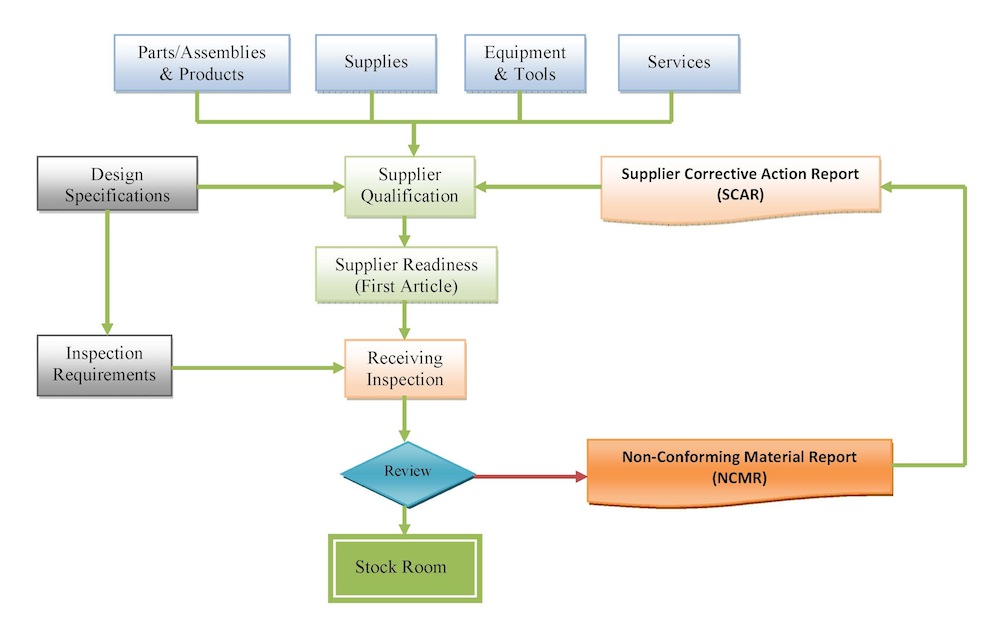
Production and Process Control
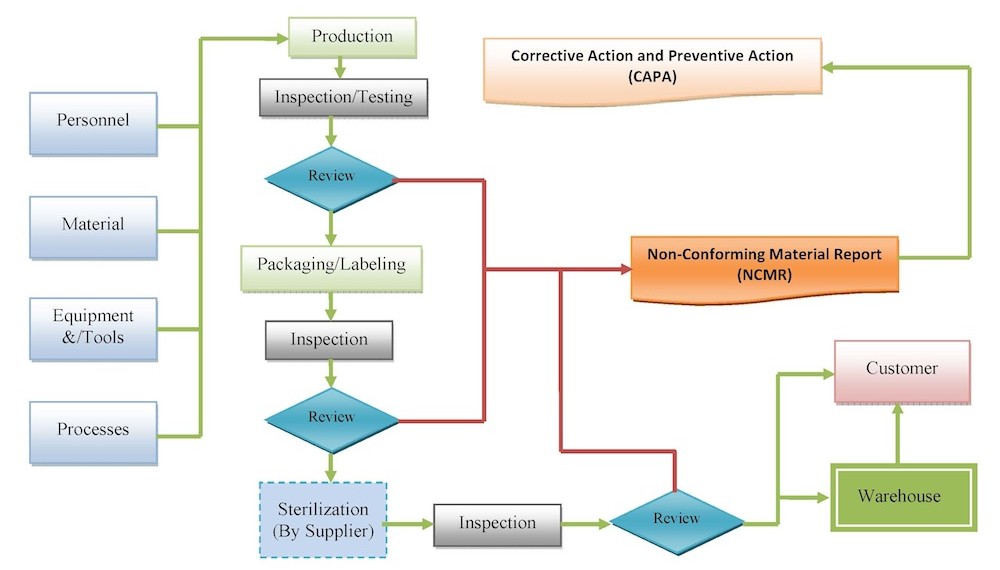
At Tyche MedTech, Inc., we ensure manufacturing consistency and repeatability in processes and products to meet established requirements by developing and implementing appropriate production and process requirements and control for the manufacture and distribution of our products. Also, we ensure that developed processes are adequate for producing products that meet specifications, and that the processes are validated (or results of those processes are fully verified), monitored and controlled. These steps help assure that resultant products meet specifications. Process control includes approved written instructions.
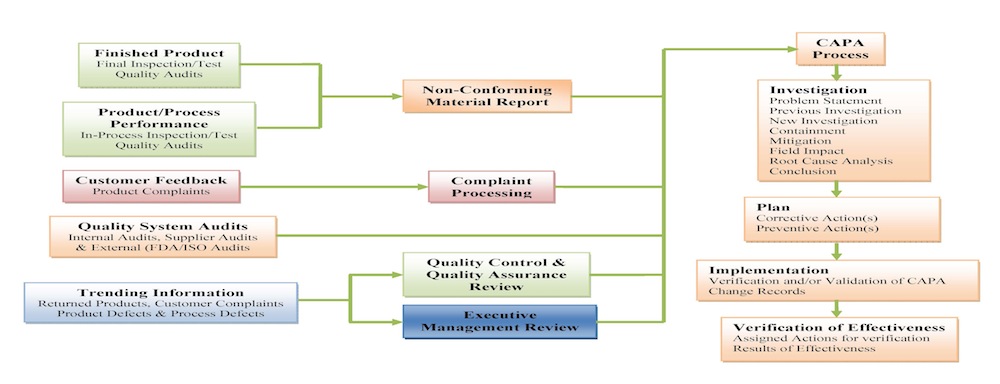
Complaint Processing
At Tyche MedTech, Inc., we pay attention to each and every complaint which may be a written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a product after it is released for distribution. Also, we review, evaluate, and, when appropriate, investigate complaints, establish and maintain written procedures describing the process used to perform these activities, and designate a responsible individual or entity to perform these tasks.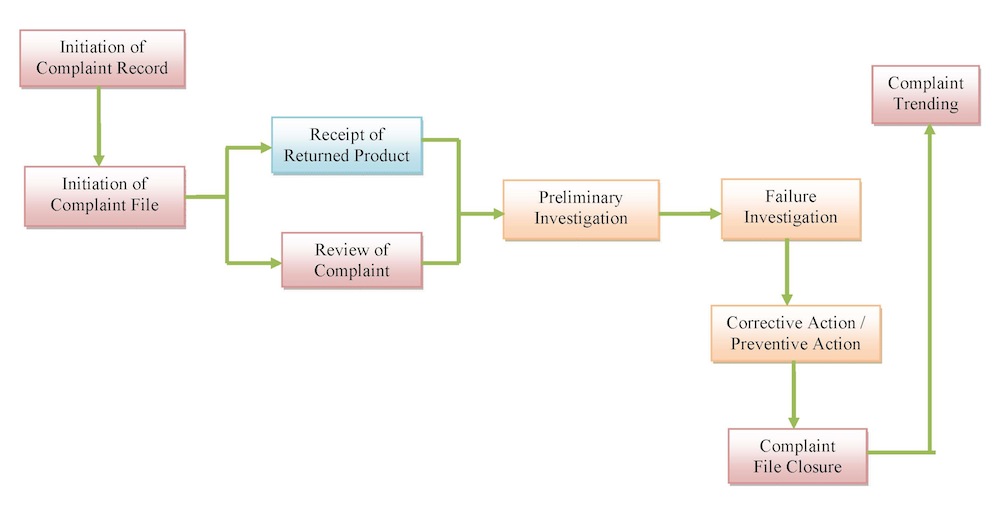
Adverse Event Reporting
At Tyche MedTech, Inc., we identify complaints concerning death, serious injury or malfunctions, and are required to report them to FDA as an adverse event or MDR (medical product reporting) and investigate them and take corrective action. We are required to submit an individual adverse event (MDR) report to FDA within 30 calendar days after becoming aware of a reportable death, serious injury, or malfunction. Also, we are required to report to FDA within 5 work days a reportable adverse event after: (1) becoming aware that a reportable adverse event necessitates remedial action to prevent an unreasonable risk of substantial harm to public health; or (2) becoming aware of a reportable adverse event from which FDA has made a written request for the submission of a 5-day report.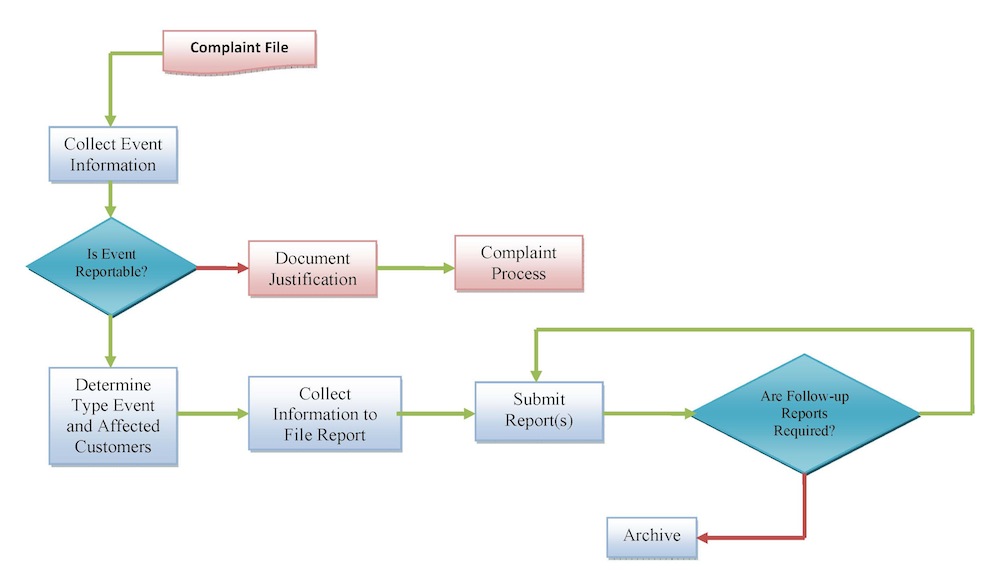
CAPA Process
At Tyche MedTech, Inc., we take corrective action to eliminate the cause of nonconformities or take preventive action to eliminate the cause of potential nonconformities in order to prevent their recurrence. We take appropriate corrective actions or preventive actions to the effects of the nonconformities encountered. For a corrective action and preventive action (CAPA) process, we have established (defined, documented and implemented) procedure that delineates requirements for: (a) reviewing nonconformities (including customer complaints), (b) determining the causes of nonconformities, (c) evaluating the need for action to ensure that nonconformities do not recur, (d) determining and implementing action needed, including, if appropriate, updating documentation, (e) recording of the results of any investigation and of action taken, and (f) reviewing the corrective action taken and its effectiveness.